Home
Products
VM2
VDock
Small Molecule Tools >
VConf
VDraw
VMap
VCharge
VFilter
Vrms
VDisplay
News
About Us
Profile
Personnel
Contact
Support
Documentation
Licensing
Science
VM2 Method
Videos
Get VeraChem Software
VDock software package
Intuitive graphical user interface (GUI) to protein-ligand docking calculation engine
Guided workflow: system setup, run docking calculations, results analysis
Seamless setup and docking direct from import of raw protein PDB file and ligand series SD file
All atom forcefield available as well as united atom
Flexible ligand and optionally flexible protein side chains
Run docking calculations directly on desktop or export calculations to HPC
Optionally, a ligand template/ligand co-crystal can define search box placement
Multiple search techniques: Global Underestimator Methods, Genetic Algorithms, Poling, and Tabu Search
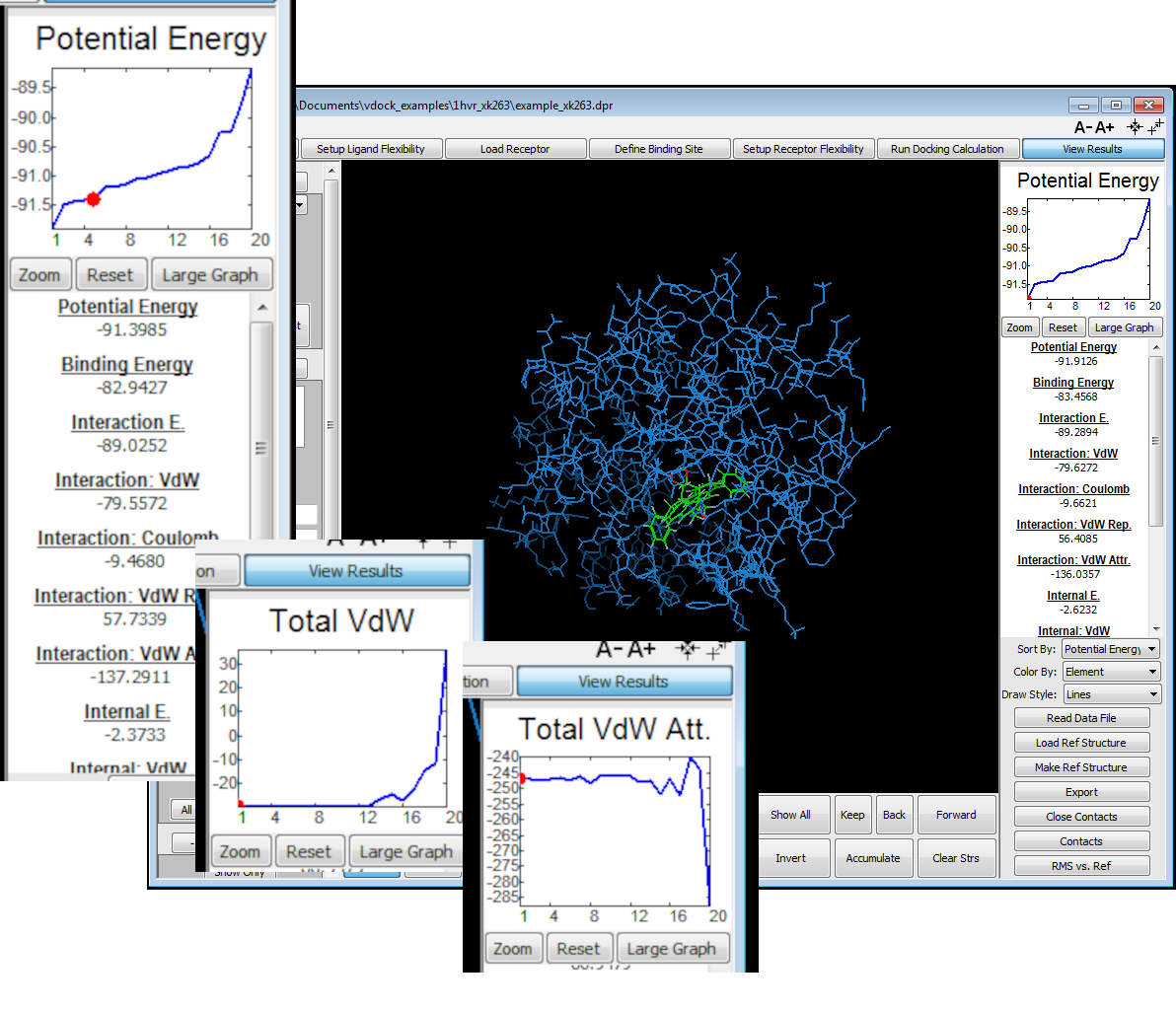
Post-docking calculation analysis, including visualization of multiple docked conformations for each ligand
Graphs of calculated energies, e.g. binding energy and its components, versus viewed conformers
Flexible commercial licensing and free for academic use cases
Get VDock
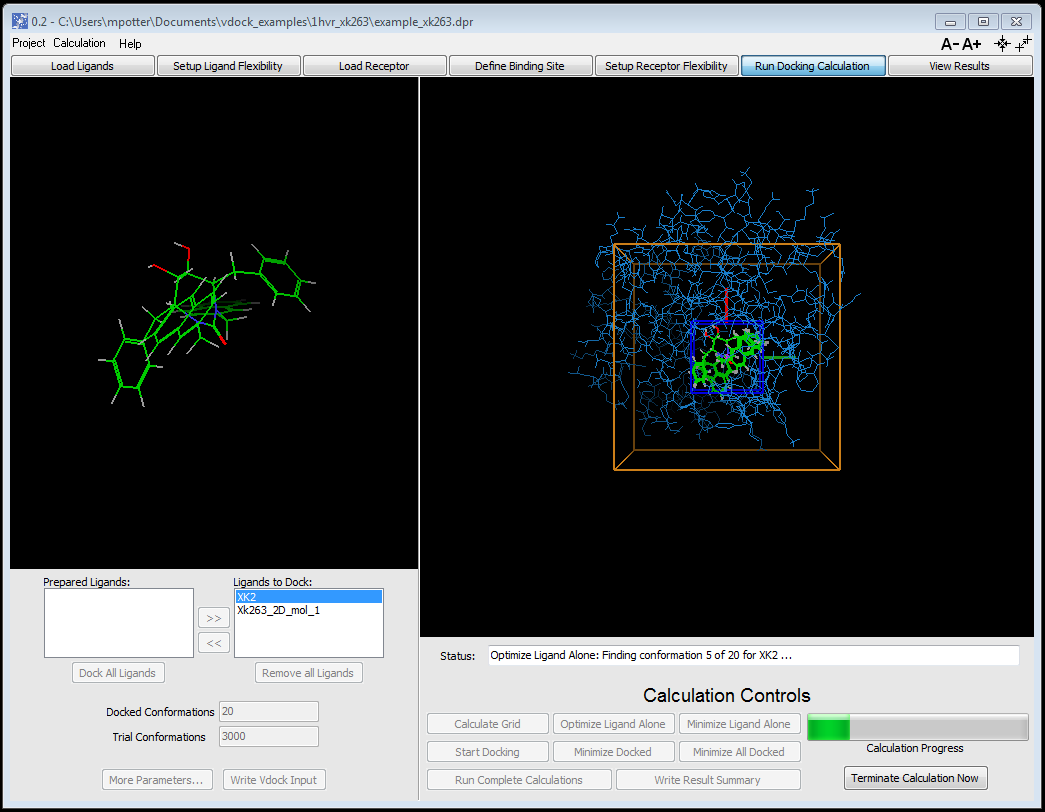
GUI based guided workflow
User guided step-by-step through system setup, calculation submission, and results analysis
Load and view target protein and ligands before and after automatic preparation and force field typing
View docked and ranked ligand conformers and graphed energies/scores
Learn More
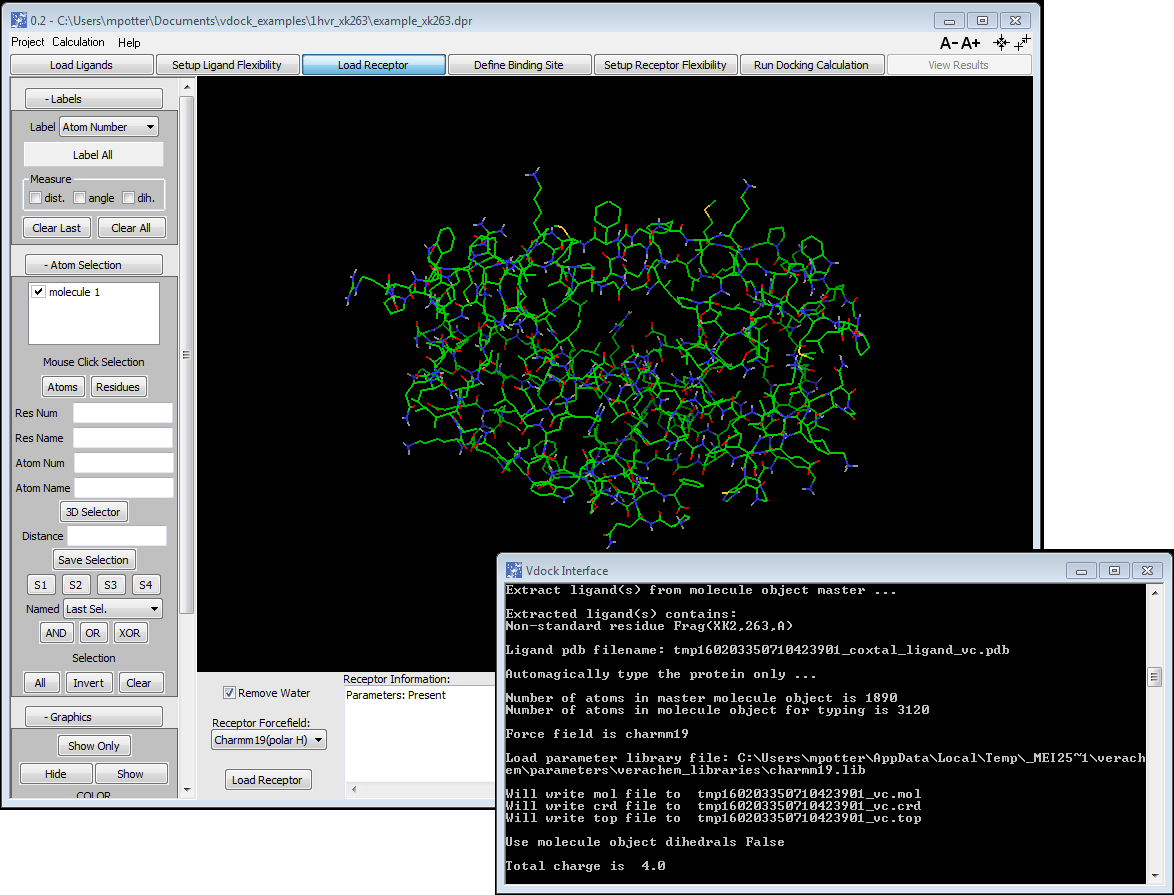
System setup direct from PDB/SDF
Import protein PDB file, prepare structure, and carry out force field typing
All atom and united atom protein forcefield typing/parameter assignment available
Enhanced Dreiding typing/parameter assignment for ligands
Learn More
Flexible ligand and protein side chains
Ligand flexibility by allowing ligand torsion angles to change
User can select protein side chains to have changeable torsion angles introducing receptor mobility
Option to fully optimize all ligand degrees of freedom after docking
Learn More
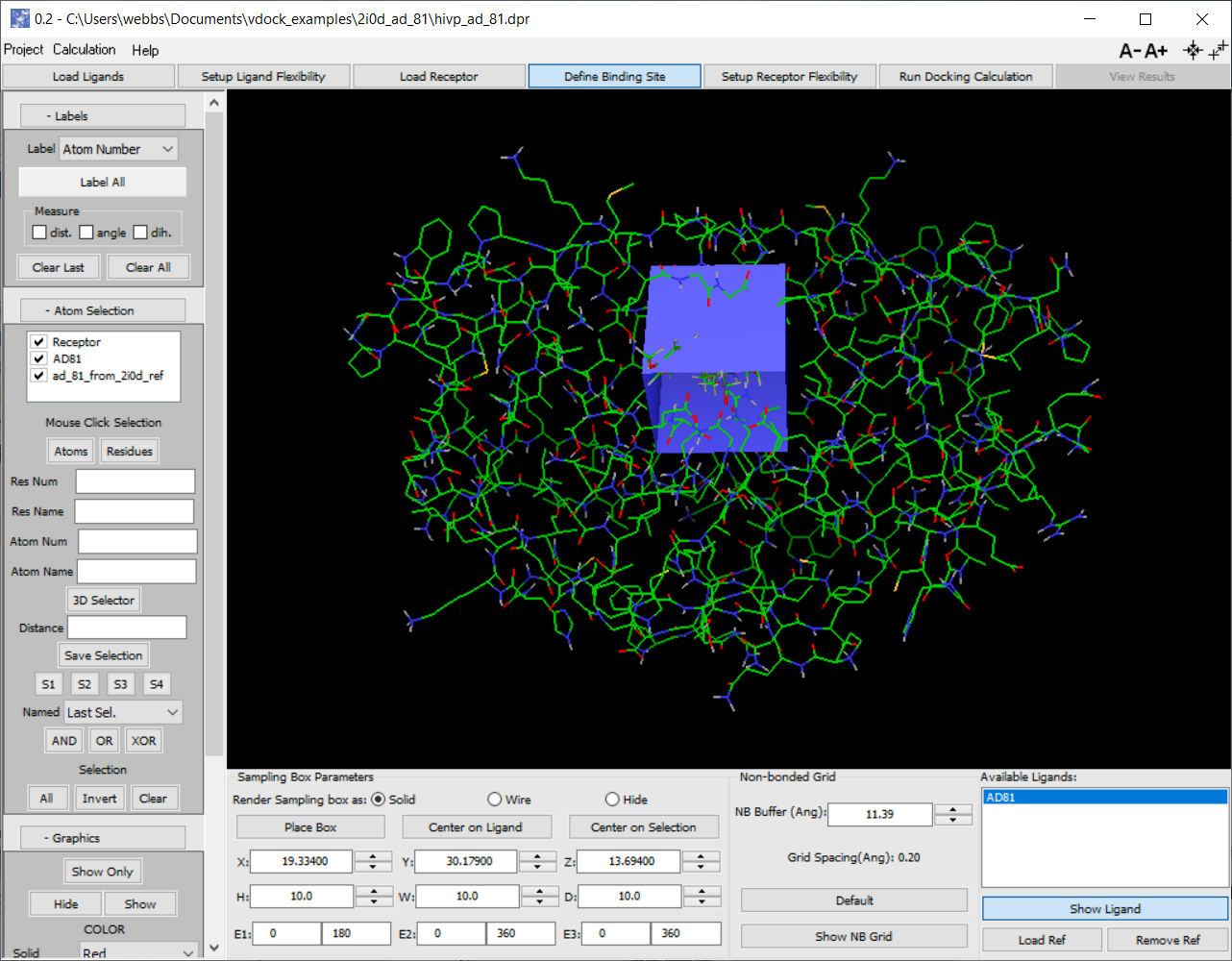
Defining the binding site
User placement and sizing of box that delimits search region
If ligand co-crystal or other reference ligand uploaded can center box on it
Learn More
Analysis of docked ligand conformers
Display multiple docked conformers for each ligand
RMSD of docked conformers with reference ligand
Interrogate ligand conformer interactions/hydrogen bonding with surrounding residues
Learn More
Ligand ranking and energy analysis
Ligands can be ranked according various metrics e.g. binding energy, interaction energy, coulomb energy, van der waals etc.
Screen large ligand sets by off-loading docking to HPC and loading results for analysis
Use entirely interactively for small to medium ligand sets – fast feedback can help guide early ligand design decisions
Learn More
Documentation
VDock Manual
VDock Tutorial